



- European Medicines Agency approves second trastuzumab and third insulin glargine biosimilars
- FDA has not approved any biosimilar drug in 2018
- Only three of nine approved biosimilars have launched in the United States
As pharmaceutical drug costs attract increasing media attention and political scrutiny, a growing number of biosimilar drugs are set to enter the U.S. and European markets in the coming years. Global sales for the top nine branded biologic drugs were estimated to total $63 billion in 2016[1]. Competition in the heavily regulated marketplace for these blockbuster therapeutics is expected to substantially impact the pharmaceutical industry and national health systems. To date, the U.S. has considerably lagged behind Europe’s expansion of biosimilar drug options. The RAND Corporation estimates that biosimilar products can save the U.S. health system approximately $54 billion over the next decade, as discussed here.
Since 2005, the biosimilar regulatory framework in Europe has been implemented through the Committee for Medicinal Products for Human Use (CHMP) under the European Medicines Agency (EMA). The CHMP provides initial assessments for marketing authorization of new medicines that are ultimately approved centrally by the EMA. Since Sandoz’s somatotropin biosimilar Omnitrope® was first authorized on April 12, 2006, an additional 39 out of 44 applications have been approved in Europe. Three of the authorizations have been withdrawn post-approval by the marketing authorization holders (Table 1).
The U.S. did not implement a regulatory framework for biosimilar evaluation until after enactment of the Biologics Price Competition and Innovation Act (BPCIA) of 2009. Given that the first U.S. biosimilar drug was approved almost a decade after the first in Europe, the number of authorized biosimilar drugs in Europe far exceeds the number of biosimilars approved in the United States. Sandoz’s filgrastim biosimilar Zarxio® received the first U.S. approval in 2015, whereas nine filgrastim biosimilars have been approved in Europe dating back to multiple authorizations in 2008. Zarxio® (in the U.S.) and Zarzio® (in Europe) are biosimilar to the reference product Neupogen® marketed by Amgen and originally licensed in 1991. Subsequent to Zarxio®’s approval, only eight other biosimilar drugs have gained U.S. approval to date (Table 2). As illustrated in the following graph, the EU’s significant head start led to the existent imbalance in the number of biosimilar drugs available in the respective markets.
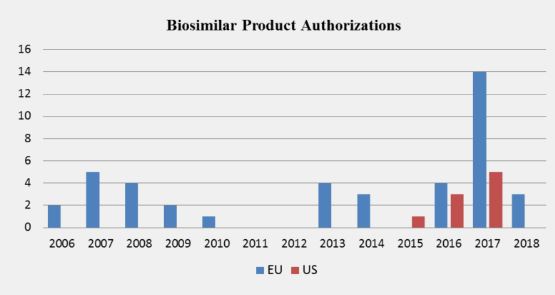
Currently, sixteen biosimilar applications are under review by the EMA for marketing authorization (Table 3). As an increasing number of U.S. patents expire on blockbuster biologic drugs, the number of abbreviated biologics license applications is also increasing. Biosimilars for at least twelve different original biologics are currently navigating the FDA’s biosimilar pathway or are in late stage development (Table 4). Interestingly, Neulasta® (pegfilgrastin) biosimilars have failed to gain approval in Europe and in the United States despite numerous attempts. The FDA has rejected pegfilgrastim applications filed by Coherus, Mylan/Biocon, and Sandoz, and there are currently eight pending applications for pegfilgrastim pending in Europe.
On February 9, 2018, the EMA approved HerzumaTM, the second trastuzumab biosimilar to Roche’s Herceptin® in Europe. The EMA previously approved Samsung Bioepis’ Ontruzant TM in November 2017. Herzuma is the third biosimilar from Celltrion’s portfolio approved by the European Commission.
On April 23, 2018, Pfizer announced that the FDA had issued a Complete Response Letter (CRL) highlighting the need for additional information for its trastuzumab biosimilar referencing Herceptin®. On April 5, 2018, Celltrion announced that the FDA had issued CRLs for 2 of its products, CT-P10, a proposed rituximab biosimilar referencing Rituxan®, and CT-P6, a proposed trastuzumab biosimilar referencing Herceptin®. The FDA has not approved any biosimilars in 2018.
Given biosimilar applicant experience in navigating the EMA process and the EMA’s high authorization rate, the FDA will need to continue to provide useful guidance and streamline the approval process in order to reduce the imbalance.
Click here to view the tables.